
 |
 |
Thrombotic microangiopathy
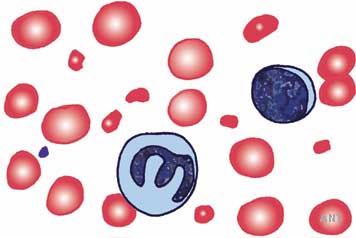
These diseases are associated with platelet thromboses in small blood vessels and consequent thrombocytopaenia. Damage to endothelium is a common factor. Other coagulation tests are not usually abnormal. Blood films show red cell fragments thought to be a consequence of damage in small vessels (microangiopathic haemolytic anaemia, MAHA; see diagram to the left) There is considerable overlap, but two major clinical patterns are recognised.
- In Thrombotic Thrombocytopaenic Purpura (TTP) haematological and central nervous system manifestations are prominent, renal disease not usually as severe. Deficiency of von Willebrand protease has been identified in some examples of TTP.
- In Haemolytic Uraemic Syndrome (HUS) renal disease is prominent. Most commonly occurs in association with bloody diarrhoea caused by verotoxin-producing E.coli (D+ HUS). In other types (Atypical, or D- HUS) endothelial damage is caused by other mechanisms. Familial susceptibility may be associated with complement abnormalities (e.g. partial deficiency of Factor H); in some of these, recurrence after renal transplantation is common.
Causes of HUS/TTP:
- Pre-eclampsia/ post-partum
Graft versus host disease after bone marrow transplantation
Drugs – quinine, cytotoxic agents, others
Verotoxins produced by some E.coli (e.g. O157) - Inherited – complement abnormalities
- Malignant hypertension may cause a clinically similar picture, and the appearance of glomerular capillaries on renal biopsy is also similar. However larger vessels often show hypertensive changes too. The renal lesion of systemic sclerosis is identical. Patient info on systemic sclerosis (scleroderma).
Disseminated intravascular coagulation (DIC)
Consumption of coagulation factors leads to thrombosis and haemorrhage in microvasculature. Management involves maintaining haemostasis, replacing clotting factors if essential and treating the underlying condition.
Myeloma and B cell dyscrasias (dysproteinaemias) – may affect the kidney in several ways:
- Hypercalcaemia
- Cast nephropathy – ‘myeloma kidney’
- Amyloidosis (AL amyloidosis caused by deposition of free light chains, see below)
- Deposition diseases (rare, caused by deposition of immunoglobulin fragments)
Myeloma is a B cell neoplasm in which there is usually overproduction of a monoclonal immunoglobulin. It causes lytic bone deposits and hypercalcaemia, bone marrow suppression, and if free light chains are overproduced they can precipitate in tubules and be toxic to tubular cells (cast nephropathy). Renal failure may be the first manifestation. Characteristically it occurs after exposure to radiological contrast media, but more often renal impairment is identified during investigation of non-specific symptoms. As in other tubulointerstitial disorders, clinical findings are minimal. Urine analysis by immunoelectrophoresis or other tests for ‘Bence Jones’ protein are required.
Amyloidosis is caused by highly structured, fibrillar aggregation of certain proteins, either:
- normal proteins present in excess, or
- proteins of abnormal sequence, giving rise to uncommon genetic causes
This leads to deposition in various tissues according to the protein, but the kidney is affected by the common types. The deposits take up the histological staining reagent Congo Red, with a characteristic apple-green colour under polarised light. Three common types of amyloid in renal patients:
- AL amyloid, caused by excess production of some immunoglobulin light chains in myeloma or in a monoclonal B cell proliferation short of overt myeloma.
- AA amyloid, caused by excess production of serum amyloid A (SAA) protein in chronic infections or inflammation, such as long term arthritis. SAA is an ‘acute phase reactant’ like C-reactive protein (CRP) in inflammation.
- Dialysis amyloid, caused by reduced excretion of beta-2 microglobulin. Exclusive to dialysis patients, in whom it causes joint and musculoskeletal symptoms.
